Institute of Bioinformatics Münster
Usage of nanopipe webtool
Start the Application
Click on "Run the Pipeline" to start the analysis. The input mask
will look like this:
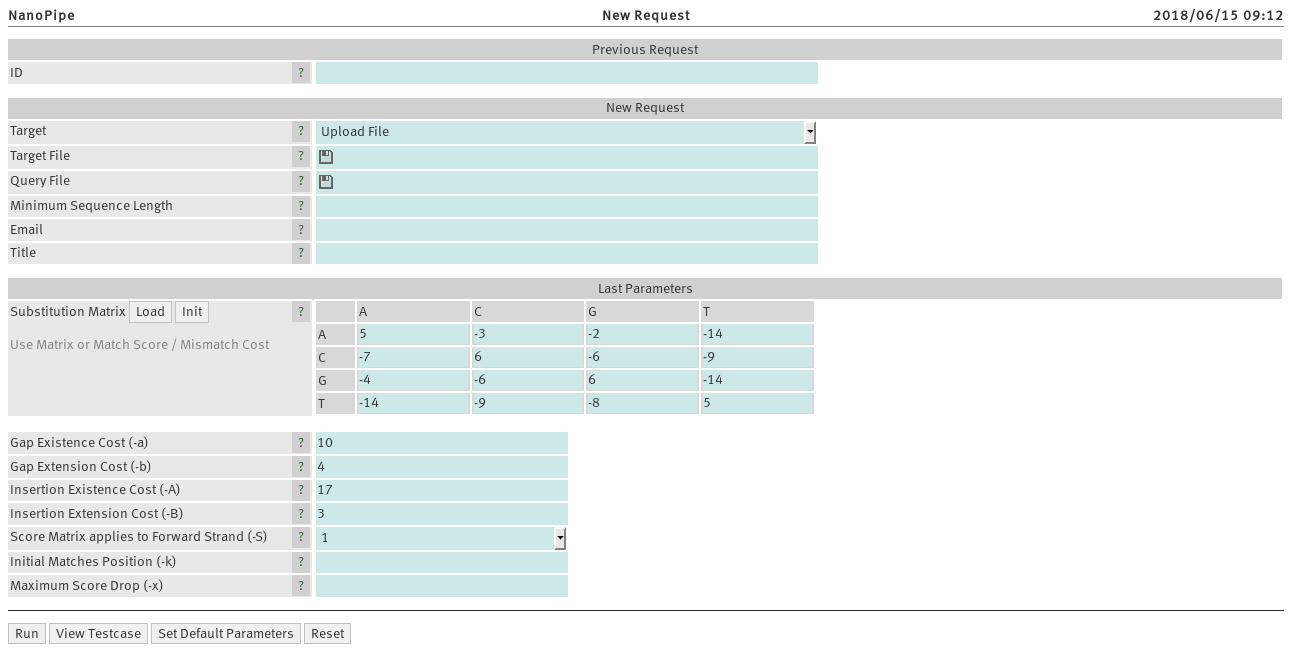 Every request is associated with a unique ID number. All running
processes have their specific ID and are handled in order of the
submission time. The last 10 request IDs are displayed in "Previous
Runs / Views" on the left side menu. One can see the previously run
processes either via this list, or by entering the request ID in
"ID" field of the input mask.
Every request is associated with a unique ID number. All running
processes have their specific ID and are handled in order of the
submission time. The last 10 request IDs are displayed in "Previous
Runs / Views" on the left side menu. One can see the previously run
processes either via this list, or by entering the request ID in
"ID" field of the input mask.
Notes
- The calculation of the results may take minutes up to hours. As long as the calculation is in process, you are redirected to a "waiting" page. It updates automatically every minute. Nevertheless, you can close the "waiting" page. Nanopipe will send you an email (if previously entered) when the calculations are finished (remember your request ID!).
- Requests are kept for 30 days on the server. After this period all the data will be deleted.
- There is a limit of 100 open requests. That means that if you try to submit your data and there are 100 requests running, you will have to wait until the queue is ready to accept requests again.
- The maximum size of a query is 3GB.
Entering Data
Target and Target File
Some targets are already prepared and displayed in the Target
selection box. If you want to provide your own target, you can
upload your file.
Query File
The query is your data and always a file (in fastq or fasta format).
You can only submit ONE file at a time. To send several files in one
request, you can archive them together and send the archive file.
Archive formats are zip. To archive files do this (in general - in
Unix)
zip archive.zip file1 file2 ...or use a Windows tool like WinZip. Then you can upload the file archive.zip to Nanopipe and proceed.
Note: Transfer and calculation times will generally increase
when sending big files.
Minimum Sequence Length
Enter the minimum length of the query reads which should be
analyzed. We recommend the minimum length of 100, but it depends on
your data and aim of the experiment.
Enter an email address to receive a notification when your job is completed.
Substitution Matrix
The substitution matrix and the fields "Match Score" and "Mismatch
Cost" are mutually exclusive - meaning that you can only use one of
them. If you do not enter values (meaning empty or "0") into
the substitution matrix, you can select "Match Score" and "Mismatch
Cost" fields. Otherwise they are invisible.
Results
When the request is finished, the main results page will be
displayed first:
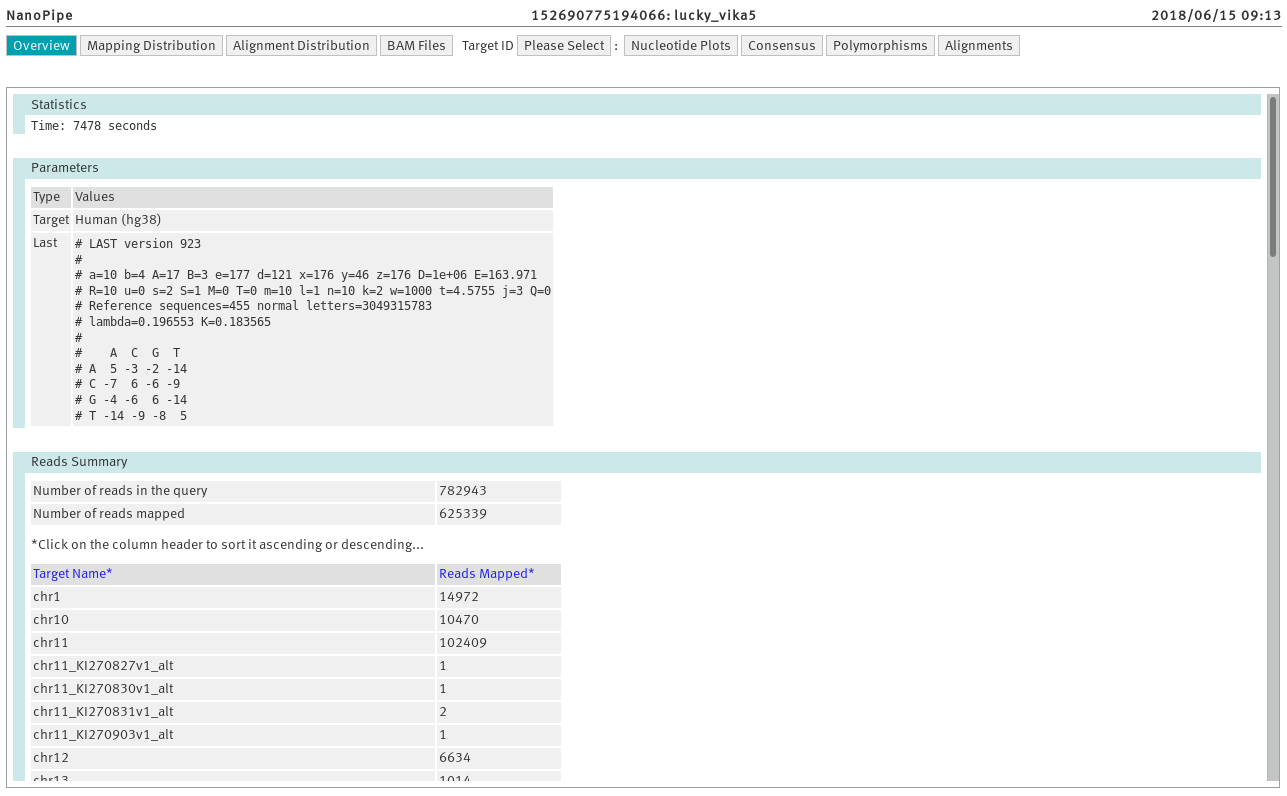 You have the option to select a specific results page, presented on
the header line (upper menu). Some of the results pages are target
name dependent: It is first required to select a
particular chromosome/contig name of the target by clicking "First
Select" before browsing through.
You have the option to select a specific results page, presented on
the header line (upper menu). Some of the results pages are target
name dependent: It is first required to select a
particular chromosome/contig name of the target by clicking "First
Select" before browsing through.
Mapping Distr
The Mapping Distribution displays the number of reads (Nreads) that
have been mapped per target.
Query Length Distr
The Query Distribution displays the distribution of the length
of the queries. Note: The graph may have a last bar which
sums up counts bigger then a specific length - this works as follows:
Try to find the length from start where 95% of all counts are occupied.
From this point go further to the right and look if the longest
length is at least twice as much as this 95% length. If so, sum
up all length together in ONE bar, otherwise display the lengths
as they are.
Alignment Length Distr
The Alignment Distribution displays the number of reads (Nreads)
with a particular alignment length (i.e., the part of the read that
has been mapped) per target.
Nucleotide Plots
The nucleotide plots display the distribution of the four
nucleotides along the target
 In the upper section you see colored bars. On the Y axis the reads
number is printed, on the X axis - the position in the
target. Vertical lines indicate a "gap" in the target position,
meaning that the read coverage at this position is less than 10. It
is possible to navigate along the graph using the menu below it. You may
move to the left or to the right, or enter an exact target
position. With "BarWidth" you can change the size of the displayed
bars. At the bottom of the Nucleotide Plot page there is an overview
graph of the plot, so you can easily find the positions with better
coverage. Just click on the alignment pick at the overview graph
and the colored nucleotide plot will move to the desired location.
In the upper section you see colored bars. On the Y axis the reads
number is printed, on the X axis - the position in the
target. Vertical lines indicate a "gap" in the target position,
meaning that the read coverage at this position is less than 10. It
is possible to navigate along the graph using the menu below it. You may
move to the left or to the right, or enter an exact target
position. With "BarWidth" you can change the size of the displayed
bars. At the bottom of the Nucleotide Plot page there is an overview
graph of the plot, so you can easily find the positions with better
coverage. Just click on the alignment pick at the overview graph
and the colored nucleotide plot will move to the desired location.
Polymorphisms
Core function
The polymorphisms table contains the information about putative
single nucleotide polymorphisms (SNP) in the analyzed sequence.
Candidates fulfill three requirements:
- The coverage of the target nucleotide is lower than or equal to 80 % of the total coverage at that position. This can be adjusted by setting the "Target threshold" during job submission. A low value will set the focus on regions which are less conserved between your query and the target. A high value will also include more conserved positions, as well.
- For non-target nucleotides at that position: The coverage must be greater than or equal to 20% of the total coverage. This can be adjusted by setting the "Polymorphism threshold" abundance during job submission. Low values will result in calls of low- and high-frequency SNPs. A high value will result in calls of only high-frequency SNPs.
-
A position on an individual target must have a certain coverage relative to
the maximum coverage of the individual target. An individual target may,
for example, be a chromosome of your target genome. By default, positions
must have a coverage of 30% of the individual target's maximum coverage.
E.g.: Chromosome 1 has a maximum coverage of 3000. Thus, positions on
chromosome 1 need to have a minimum coverage of 1000.
You can change the default by adjusting the "Coverage threshold" during job submission. Lower the value to include also low-coverage positions. Increase the value to exclude low-coverage positions.
After this filtering step, the remaining SNPs are weighted. For
that, the relative occurrence of each base at the given position is
calculated. Then, the changes from the target nucleotide to the
observed nucleotide are evaluated. The assumption here is that
transitions are twice as likely as transversions. The weighted
occurrence of each nucleotide shows the likelihood of observing a
nucleotide different from the target.
Additional information
1) The algorithm provides additional information for each SNP
candidate from dbSNP for Homo sapiens and
from PlasmoDB for Plasmodium falciparum. If matching
entries are found, the corresponding IDs and other available
information are added to the output.
Note: For human data we use the novel API of dbSNP
which allows for searches on a valid chromosome. Non-valid
chromosomes include chrM and chromosomes ending with further
comments, like: chr1_mycomment.
Non-valid target sequences are indicated by N/A in the
respective output column. db:error indicates that the
attempt to connect with dbSNP has failed.
2) As another additional feature, the alignment quality is
being analyzed: The p-value is calculated based on the LAST alignment
quality values. The average p-value for the region of 10 bases before
and 10 bases after the candidate SNP is estimated.
The interpretation of this value may be following:
- The SNP lays in a region of low alignment quality. Therefore, it is likely reflecting a sequencing error and not a biological pattern.
- The SNP lays in a SNPs cluster and this reciprocally affects the alignment quality.
The output
The output will look similar to the following example:
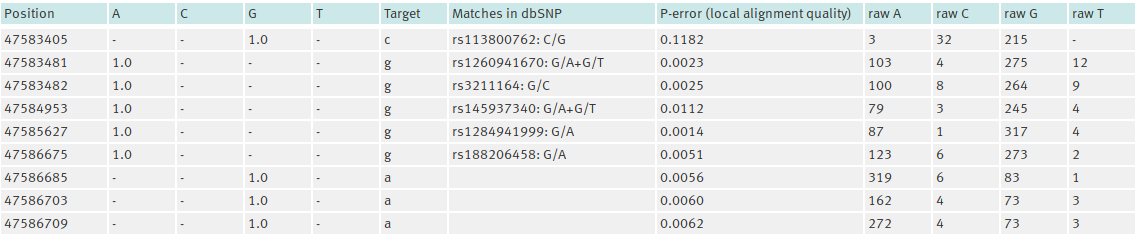 The rows of the table include the following information:
The rows of the table include the following information:
- Position: The position of the SNP candidate.
- A, C, G, T: The joint probabilities of the SNP being an A, C, G or T (see above).
- Target: The nucleotide in the target.
- Matches in dbSNP/Matches in PlasmoDB: For dbSNP: [rs-IDs: alleles] are reported for the position. db:error indicates a failed try to contact dbSNP. For PlasmoDB: [ID: major allele: frequency + minor allele: frequency] are reported. No entry means no match, N/A indicates that a chromosome is not in the databases (see above). The SNP data base data are currently only provided for the two species: H. sapiens and P. falciparum. For any other target types, this column is missing.
- P-error (local alignment quality): The average p-value around a SNP (see above).
- raw A, raw C, raw G, raw T: Raw coverage observed for A, C, G and T.
Consensus
The consensus of the sequenced DNA or RNA is provided. It is
generated based on the majority rule, i.e. the nucleotide with the
highest count at a particular position is being assigned to the
consensus sequence at this position. If a nucleotide has coverage of
less than 10, a gap is introduced. If the nucleotides' coverages at a
position are similar (within 80% similarity), NanoPipe uses the IUPAC
nomenclature to acknowledge this uncertainty,
see IUPAC
code.
Note: in the consensus sequence "X" stays for any of the four
nucleotides, and "N" meaning that there were not enough data to make
a decision.
Alignments
The pairwise alignment of consensus and target sequences is displayed
here.
BAM Files
We generate .bam and .bai files for you, so you could visualize the
results in IGVviewer
(see Broadinstitute). The
FASTA file with the corresponding target sequence is also available
for download, as it is required for IGVviewer.
2020-09-08 09:48